

こんにちは、eURO2023です!
趣味(?)でRstudioをしています。
real-time PCRの結果を、Rstudioで作成してみたので備忘録としてまとめておきます。
このreal-time PCRについては、Excelで十分作れてしまうのでか、あまりRstudioでやったという記事がないんですよね…。
実際にChatGPTの助けも借りながら、スクリプトを作成してみました。
思ったより煩雑なスクリプトとなってしまいましたが、グラフは割とキレイに作成できたので、気になる方は参考にしてみてください!
スクリプト全景はこちらを確認してください。
医師が統計を学ぶ上で、絶対読んだほうがよい!と思われる書籍を上げておきます。


ファイル読み込み or 架空のCqデータシート作成
恒例の、架空データシート作成です。
# ライブラリの読み込み
library(dplyr)
library(ggplot2)
library(scales)
# 変数の定義
samples <- c("A", "B", "C", "D", "E", "F")
target_genes <- c("geneA", "geneB", "geneC")
reference_gene <- "GAPDH"
replicates <- 3
# 再現性のためのシード設定
set.seed(333)
# 架空データシートの作成
df <- expand.grid(Sample = samples,
Target = c(reference_gene, target_genes),
Replicate = seq_len(replicates)) %>%
mutate(mean_Cq = case_when(
Target == reference_gene ~ 20,
Sample == "B" & Target != reference_gene ~ 28,
TRUE ~ 22),
Cq = rnorm(n(), mean = mean_Cq, sd = 0.2)) %>%
dplyr::select(-mean_Cq, -Replicate)
# データの表示
head(df)
#------------------
## CSVファイルを読み込む場合
df <- read.csv("読み込みたいファイルの絶対パス", stringsAsFactors = FALSE)
head(df)一つずつみていきましょう!
ライブラリの読み込み
library(dplyr)
library(ggplot2)
library(scales)dplyr はデータ操作を簡単に行うためのライブラリです。
mutate, case_when, filter, group_by, summarise, left_join, ungroup, rowwise, などのコマンドを使用するためにライブラリを読み込んでおきます。
ggplot2は、キレイなグラフを作成するためには(私の考えでは)必須のパッケージです!!
scalesは、グラフの色を設定するために読み込んでおきます。
RColorBrewerがよく使われるパレットですが、scalesを用いたカラー設定は、かなり多くの色を柔軟に設定可能なので、今回はscalesを用いようと思います。
変数の定義(自分のデータを読み込む場合にも必須)
samples <- c("A", "B", "C", "D", "E", "F")
target_genes <- c("geneA", "geneB", "geneC")
reference_gene <- "GAPDH"
replicates <- 3samples :細胞の種類(A, B, C, D, E, F…)
targets :ターゲット遺伝子(geneA, geneB, geneC…)
reference_gene :使用するハウスキーピング遺伝子(reference gene)を入力
replicates :それぞれの組み合わせごとのリピート回数(3回)
samples, targets, replicatesはそれぞれ自由に変更できます!
samplesは2個以上(1個でもグラフはかけますが、実際の実験で1つであることは無いでしょう)で自由に設定できます。
targetsは1つ以上で設定できます。
reference_geneは今回「GAPDH」にしていますが、β-actinや他のハウスキーピング遺伝子でも問題ありません。
replicatesは1から設定可能です。殆どの場合はduplicate以上で実験が行われるものと思います。
この設定は、皆さんのデータをCSVやexcelファイルから読み込む場合にも用いるため、必須の設定となります!
架空のデータシート作成(自分でデータを持っている人は不要)
##架空のデータシート作成
# 再現性のためのシード設定
set.seed(333)
# 架空データシートの作成
df <- expand.grid(Sample = samples,
Target = c(reference_gene, target_genes),
Replicate = seq_len(replicates)) %>%
mutate(mean_Cq = case_when(
Target == reference_gene ~ 20,
Sample == "B" & Target != reference_gene ~ 28,
TRUE ~ 22),
Cq = rnorm(n(), mean = mean_Cq, sd = 0.2)) %>%
dplyr::select(-mean_Cq, -Replicate)
# データの表示
head(df)再現性のためのシード設定
set.seed(333)set.seed() は、乱数を固定して毎回同じデータを生成できるようにするためのものです。
これを設定しないと、スクリプトを実行するたびに異なるCq値が生成されます。
()内は自分の好きな番号に設定してください!
一応、ここでは「333」にしていますが、何となくです(笑)
架空データシートの作成
df <- expand.grid(Sample = samples,
Target = c(reference_gene, target_genes),
Replicate = seq_len(replicates)) %>%expand.grid() は、全ての組み合わせを作成する関数です。
ここでは、先ほど定義した変数:
samples :A, B, C, D, E, F
targets :geneA, geneB, geneC
reference_gene :GAPDH
これら変数を用いて、すべての組み合わせを作成します。
例えば上記のように6つのサンプル、4つの遺伝子(1つのreference_gene + 3つのtarget_genes)がある場合、6×4 = 24行のデータフレームがまず作成されます。
次に
Replicate = seq_len(replicates))
で、指定したreplicatesの数だけ、上記データフレームが複製されます。
つまり、結果として6×4×3 = 72行のデータフレームが生成されます。
★ここで注意点ですが、samplesの先頭にくるサンプル(ここではA)が、今後のrelative expression計算において基準サンプルになります。
基準サンプルを変更したい場合は、基準サンプルを先頭に持ってくるようにしてください!
mutate(mean_Cq = case_when(
Target == reference_gene ~ 20,
Sample == "B" & Target != reference_gene ~ 28,
TRUE ~ 22),
Cq = rnorm(n(), mean = mean_Cq, sd = 0.2)) %>%mean_Cq = case_when(...)
- 各行において「Cq値の平均値」を仮定して決定します。
- 基準遺伝子(GAPDH):すべて20に設定(正規化用)
- SampleがBで、ターゲット遺伝子(GAPDH以外):28に設定(発現が低い想定)
- それ以外:22(通常の発現)
ここは適当で良いですが、できるだけ最後に有意差をつけたいので、sample Bと他の発現を区別しました。
rnorm((n(), mean = mean_Cq, sd = 0.2))
上で指定した平均 mean_Cq に基づいて、正規分布のランダムなCq値を生成します。
rnorm()は正規分布(ガウス分布)に従う乱数を生成する関数です。
nは生成する乱数の個数、meanは正規分布の平均値を示します。先程設定したmean_Cqに習うように設定しています。
sdはstandard deviation、標準偏差です。サイクル数のばらつきを模す(測定誤差)ためにつけます。
今回は”ばらつき”を小さくしたい(できるだけ有意差をつけたい)、ため、sd = 0.2としました。
dplyr::select(-mean_Cq)最後に、実際のデータではmean_Cqは無いと思いますので、消します。
Replicateがデータシートに出てくることはあまり無いのですが、relative expressionの計算に必要になってくるので、残します。
select(-“消したい列”)で、消去したい列だけを削除できます!
データの表示
> head(df)
Sample Target Replicate Cq
1 A GAPDH 1 20.28664
2 B GAPDH 1 22.68572
3 C GAPDH 1 20.21320
4 D GAPDH 1 19.84843
5 E GAPDH 1 20.29972
6 F GAPDH 1 22.13254架空のreal-time RT-PCRのデータシートを作成できました!
print(df)でも良いのですが、全てのデータが表示されてしまうので、面倒くさいです。
head(df)にすれば、最初の6行のみが表示されます!
## CSVファイルを読み込む場合
df <- read.csv("読み込みたいファイルの絶対パス", stringsAsFactors = FALSE)
head(df)
#読み込んでから変数を設定しても良い
samples <- c("A", "B", "C", "D", "E", "F")
target_genes <- c("geneA", "geneB", "geneC")
reference_gene <- "GAPDH"
replicates <- 3自身のデータを用いたい場合には、read.csv関数を使って読み込みましょう。
自身のデータを用いる場合も、先程最初に設定した変数の定義は必要です。
読み込んだ後に、上記のように設定されても構いません。
Replicate列が無い場合は作成しておきましょう。
また、それぞれの列もSample, Target, Replicate, Cqと整合性をあわせておいてください。
準備は終了しました、いよいよデータ成形、グラフの作成です!
ΔΔCt値の取得
以降のスクリプトは、そのままコピペして使っていただいても成り立つと思いますが、順に説明していきます。
# reference gene の Cq を Replicate ごとに抽出
reference_df <- df %>%
filter(Target == reference_gene) %>%
dplyr::select(Sample, Replicate, reference_Cq = Cq)
# 他のターゲット遺伝子と結合し、ΔCt を計算
df <- df %>%
filter(Target != reference_gene) %>%
left_join(reference_df, by = c("Sample", "Replicate")) %>%
mutate(dCt = Cq - reference_Cq)
# 基準サンプル(samples[1])の ΔCt 平均(Target ごとに)
baseline_df <- df %>%
filter(Sample == samples[1]) %>%
group_by(Target) %>%
summarise(baseline_dCt = mean(dCt, na.rm = TRUE), .groups = "drop")
# ΔΔCt と Relative Expression を計算
df <- df %>%
left_join(baseline_df, by = "Target") %>%
mutate(
ddCt = dCt - baseline_dCt,
Relative_Expression = 2^(-ddCt)
)reference geneのCqをReplicateごとに抽出
reference_df <- df %>%
filter(Target == reference_gene) %>%
dplyr::select(Sample, Replicate, reference_Cq = Cq)まずdf内からfilter()で各サンプルのreference gene(今回はGAPDH)のみに絞ります。
この中からCq値をreference_Cqとして保存します。
dplyr::select()でReplicateごとのCqを保持しておくことで、ΔCt計算時にペアで比較可能になります!
ちなみにselect()でも良いのですが、select()コマンドは多くのパッケージに含まれているので、無駄なエラーメッセージを避けるためにもdplyr::select()と明示してあげると良いです(dplyrコマンド内の、selectを使いますよと明示する)。
他のターゲット遺伝子と結合し、ΔCtを計算
df <- df %>%
filter(Target != reference_gene) %>%
left_join(reference_df, by = c("Sample", "Replicate")) %>%
mutate(dCt = Cq - reference_Cq)df には「GAPDH(基準遺伝子)」とターゲット遺伝子(geneA, geneB, geneC)の すべてのCq測定値 が含まれています。
filter(Target != reference_gene)ΔCtの計算はターゲット遺伝子に対してだけ行うので、GAPDH(reference gene)はここで取り除きます。
Target != reference_geneで、reference_gene(この場合は、GAPDH)のみをTarget列から除くことができます。
left_join(reference_df, by = c("Sample", "Replicate"))先ほど処理した通り、reference_dfには各サンプル×リプリケートに対応する GAPDHのCq値(reference_Cq)が入っています。
これを、lelft_join()を使うことによって 同じ Sample かつ 同じ Replicate の行に結合して、
- Cq(ターゲット遺伝子のCq値)
- reference_Cq(基準遺伝子のCq値)
の ペアデータを1行に揃えることができます。
ここで、最初に設定したReplicate列が重要になってきます。
- 例えば Sample “A” の replicate 1 の gene A 測定は、Sample “A” の replicate 1 の GAPDH とだけ対応します。
- Replicateを無視すると、対応がズレて誤ったΔCtが計算されてしまいます。
mutate(dCt = Cq - reference_Cq)それぞれのターゲット遺伝子について、ΔCt を計算します。
ΔCt=ターゲット遺伝子のCq – リファレンス遺伝子のCq
この差をとることで、ターゲット遺伝子の発現量を基準遺伝子で正規化することが出来ます!
基準サンプル(samples[1])のΔCt平均(Targetごとに)
baseline_df <- df %>%
filter(Sample == samples[1]) %>%
group_by(Target) %>%
summarise(baseline_dCt = mean(dCt, na.rm = TRUE), .groups = "drop")続いて、ΔΔCt値を測定するために、基準サンプルのΔCt値のみを抽出します。
filter(Sample == samples[1])sample[1]は、定義されたサンプルベクトルの最初の要素です。
今回はsamples <- c(“A”, “B”, “C”, “D”, “E”, “F”)
となっているので、sample[1]は”A”となります。
基準サンプルを変更したい場合は、このスクリプトを変更するか(sample[2]とすれば、左から2番目の要素が基準サンプルになる)、最初のベクトル定義を変更してください。
ただ、このスクリプトは自動で最後まで進めるようになっているので、ベクトル定義を変更していただくほうが後々迷わなくていいかもしれません。
group_by(Target)“A”のデータの中で、各遺伝子(geneA, geneB, geneC)ごとにグループ化します。
このあとmean()を使って、各遺伝子のΔCt の平均を求める準備になります。
summarise(baseline_dCt = mean(dCt, na.rm = TRUE), .groups = "drop")mean(dCt, na.rm = TRUE):各遺伝子について "A" の replicate(複数回測定)から ΔCt の平均を取ります。
na.rm = TRUE は、もし欠損値(NA)があっても無視して平均を取る指定です。
つまり、replicate=3で、うち一つにNAがあった場合、N=2で平均を取ってくれるようにしています。
.groups = "drop":group_by() の影響をここで解除しています。
ΔΔCtとRelative Expressionを計算
df <- df %>%
left_join(baseline_df, by = "Target") %>%
mutate(
ddCt = dCt - baseline_dCt,
Relative_Expression = 2^(-ddCt)
)さて、いよいよ相対発現量(Relative Expression)を求める処理にうつります!
left_join(baseline_df, by = "Target")先ほど作成した baseline_df(基準サンプルAの各遺伝子における平均ΔCt)を、全データに結合します。
baseline_dfにはTarget列とbaseline_dCt列があります。dfのTargetに対応するbaseline_dCtがdfに加えられることになります。
mutate(
ddCt = dCt - baseline_dCt,
Relative_Expression = 2^(-ddCt)
)このスクリプトで、ΔΔCtとrelative expressionをまとめて計算します!
各サンプルの ΔCt から、基準サンプルの ΔCt(baseline_dCt)を引いて、ΔΔCtを計算(ddCt = dCt – baseline_dCt)します。
つづいて、得られたddCtを元に2^(-ddCt)でrelative expressionを計算します(”^”は乗を表しています)。
さて、ここから統計学的検定とグラフの作成にうつっていきます!
t検定
PCRの検定には皆さん何を用いられていますか?
おそらく、よくt検定が用いられているものと思います。
しかし、基本的にt検定は正規性がある場合(パラメトリック)、かつ2群間の比較に用います。
one-way ANOVAが用いられているケースもよく見かけますが、これもパラメトリック検定です。
3群間以上、かつ正規性にとらわれない解析方法の一つとしてはKruskal-Wallis検定があり、これで有意差があった場合は多重比較をDunn検定で行う…というのが、おそらくは真っ当な方法と思われます。
しかし実際Kruskal-Wallis検定が行われている論文はあまり見かけず、t検定やone-way ANOVAが多いように思われます。
ここではt検定について、下記にプロトコルを載せますが、後日Kruskal-Wallis検定についても改めて記載しようと思っています。
一つずつ見ていきましょう!
t検定→有意差ありのペアのみ抽出
t_test_res <- df %>%
group_by(Target) %>%
summarise(pmat = list(pairwise.t.test(Relative_Expression, Sample, p.adjust.method = "BH")$p.value), .groups = "drop") %>%
mutate(pmat = map(pmat, ~ as.data.frame(.x) %>%
tibble::rownames_to_column("Sample1"))) %>%
dplyr::select(Target, pmat) %>%
unnest(pmat) %>%
pivot_longer(cols = -c(Target, Sample1), names_to = "Sample2", values_to = "p_adj") %>%
filter(!is.na(p_adj) & p_adj < 0.05) %>%
mutate(signif = case_when(p_adj < 0.001 ~ "***", p_adj < 0.01 ~ "**", TRUE ~ "*"))ペアワイズt検定、多重比較補正
t_test_res <- df %>%
group_by(Target) %>%
summarise(
pmat = list(
pairwise.t.test(Relative_Expression, Sample, p.adjust.method = "BH")$p.value
),
.groups = "drop"
)各Target(遺伝子)ごとに、サンプル間でt検定を実行(ペアワイズ)します。
pairwise.t.test(...) はすべての サンプルの組み合わせに対して比較を行い、p値の行列(matrix)をpmatに入れて返します。
p.adjust.method = "BH" は Benjamini–Hochberg法 によるFDR補正(false discovery rate correction)を行っています。
Benjamini–Hochberg法(BH法)とは、多重検定による偽陽性を制御するための方法で、他に有名なBonferroni法などがあります。
p.adjust.method = "BH" の部分を、”bonferroni”に変えるとBonferroni法、
“holm”に変えるとHolm法、
“hochberg”にするとHochberg法…というふうに多重比較補正の方法を変更できます!
List “pmat”の調整
mutate(pmat = map(pmat, ~ as.data.frame(.x) %>%
tibble::rownames_to_column("Sample1")))先ほど作成されたpmat(matrix)を as.data.frame() に変換し、行名(元はSample名)を Sample1 という列に変換します。
map(...) によって Target ごとに繰り返し処理されます。
| Sample1 | B | C | D |
|---|---|---|---|
| A | 0.03 | 0.05 | 0.002 |
| B | NA | 0.09 | 0.01 |
| C | NA | NA | 0.07 |
pmatは上記のようなdata frameのリストになっているはずです(パイプ演算で繋いでいるのでみえませんが…)。
pmatの展開
dplyr::select(Target, pmat) %>%
unnest(pmat)ここはちょっとややこしいですが…
pmat内の情報は、dfの中の”pmat”列の中にlistとして埋め込まれています。
dfの中の列をTarget, pmatにしぼり(dplyr::select(Target, pmat) )、
unnest()コマンドにてpmat列をSample1, B, C, D…のようにtidy形式に展開します。
p値、対応する組み合わせの整理
pivot_longer(
cols = -c(Target, Sample1),
names_to = "Sample2",
values_to = "p_adj"
)pivot_longer()にて、横長のB, C, D, …をSample2列として新たに設定し、p値はp_adjとして新たな列を作成します。
| Target | Sample1 | Sample2 | p_adj |
|---|---|---|---|
| geneA | A | B | 0.03 |
| geneA | A | C | 0.05 |
| geneA | A | D | 0.002 |
| geneA | B | C | 0.09 |
| … | … | … | … |
結果として、このようなdata frameに変換されることになります。
有意差のみを残し、グラフ用にp値をアスタリスクに変換
filter(!is.na(p_adj) & p_adj < 0.05) %>%
mutate(signif = case_when(p_adj < 0.001 ~ "***", p_adj < 0.01 ~ "**", TRUE ~ "*"))グラフ作成のためのデータ成形、最終です!
グラフに記載する際、有意差のあるものだけを残しますよね?
ここでは標準的にp<0.05を有意差ありと設定し、p値の欠損しているもの(is.na(p_adj))と有意差の無いもの(p_adj < 0.05)を省きます。
そして、その中でもp<0.001を***、p<0.01を**、p<0.05を*としてグラフに記載できるようにします。
ここは各自でカスタマイズ可能です!
このアスタリスクは、singifという列に新たに作成されます。
最終的に、これらのデータはt_test_resとして新たに格納されます!
グラフ上での有意差表記
t_test_res_plot <- t_test_res %>%
group_by(Target) %>%
mutate(
top_y = max(df$Relative_Expression[df$Target == unique(Target)], na.rm = TRUE) * 1.05,
x1 = match(Sample1, samples),
x2 = match(Sample2, samples),
x_mid = (x1 + x2) / 2,
y_offset = row_number(),
y_position = top_y + (y_offset - 1) * 0.1 * top_y
) %>%
ungroup()group_by(Target)
- 各遺伝子(Target)ごとにグループ化します。
→ 最後のグラフでfacet_wrapを用い、グラフを遺伝子ごとに分けますが、各遺伝子毎に y軸の高さを決めるためです。
top_y = max(df$Relative_Expression[df$Target == unique(Target)], na.rm = TRUE) * 1.05
- 各遺伝子の中で最大の相対発現量(Relative Expression)を取得します。
- それに 1.05倍 して「アスタリスクなどを載せるための土台高さ」として設定します。
→ つまり「棒グラフより少し上」の y値を取得しています。
*1.05を*1.02などと変更することで、比較線の位置を微調整できます。
x1 = match(Sample1, samples) / x2 = match(Sample2, samples)
- Sample1・Sample2 の 棒グラフ上のx軸位置(順番)を取得します。
x_mid = (x1 + x2) / 2
- x 軸上での 比較線の中央位置(アスタリスク表示位置) を計算します。
→ グラフ作成時のgeom_text()でアスタリスクを表示するために使っています。
y_offset = row_number()
- グループ内(Targetごと)での行番号(1, 2, 3…)
- 同じ Target 内で複数の比較がある場合、ラベルが重ならないようにラベルを段違いに表示するためのオフセット量になります。
y_position = top_y + (y_offset - 1) * 0.1 * top_y
- 最初の比較(1行目)は
top_yに表示させます。 - 2つ目以降は、0.1×top_y ずつ上にずらすように設定しています(ラベルが重ならないように)
→geom_segment()やgeom_text()で線・ラベルの高さとして使います。 - このズレを調整する場合は、0.1を0.2や0.05…などと変更してください。
有意差表示をグラフ内に収めるための工夫
y_limits <- t_test_res_plot %>%
group_by(Target) %>%
summarise(max_label_y = max(y_position + 0.1 * top_y, na.rm = TRUE), .groups = "drop") %>%
mutate(Sample = samples[1])summarise(max_label_y = max(y_position + 0.1 * top_y))
- 各Targetごとに、一番上にくるアノテーションのさらに上(少し余裕をもたせる)をy_limitsとして指定します。
→ これをylim()で使うことで、比較線とアスタリスクが見切れてしまうことを防ぎます。
mutate(Sample = samples[1])
facet_wrap()の軸や補助線に使うために、x軸位置に対応するSampleを補完しています。- この表示にあまり意味はありませんが、グラフ作成時のエラーを防ぐ目的で置いてます。
データの可視化
さて、いよいよグラフ作成による可視化に移ります!
sample_colors <- setNames(hue_pal()(length(samples)), samples)
ggplot(df, aes(x = factor(Sample, levels = samples), y = Relative_Expression, fill = Sample)) +
stat_summary(
fun = function(x) exp(mean(log(x))),
geom = "bar",
color = "black",
width = 0.6,
position = position_dodge(0.7)
) +
stat_summary(
fun.data = function(x) {
m <- mean(log(x))
se <- sd(log(x)) / sqrt(length(x))
data.frame(
y = exp(m),
ymin = exp(m - se),
ymax = exp(m + se)
)
},
geom = "errorbar",
width = 0.2,
position = position_dodge(0.7),
color = "black"
) +
facet_wrap(~ Target, scales = "free_y") +
scale_fill_manual(values = sample_colors) +
labs(
title = "Relative Gene Expression (ddCt Method)",
x = "Sample",
y = "Relative Expression (2^(-ddCt))"
) +
theme_minimal(base_size = 14) +
theme(legend.position = "top") +
geom_segment(
data = t_test_res_plot,
aes(x = x1, xend = x2, y = y_position, yend = y_position),
inherit.aes = FALSE,
color = "black",
linewidth = 0.5
) +
geom_text(
data = t_test_res_plot,
aes(x = x_mid, y = y_position + 0.01 * top_y, label = signif),
inherit.aes = FALSE,
size = 5
) +
geom_blank(data = y_limits, aes(x = 1, y = max_label_y))なんだかとってもややこしいスクリプトが出てきましたが…
基本コピペしてもらったら下のようなキレイなグラフになるのですが、カスタマイズのためにも一通りスクリプトの内容を確認していきましょう!
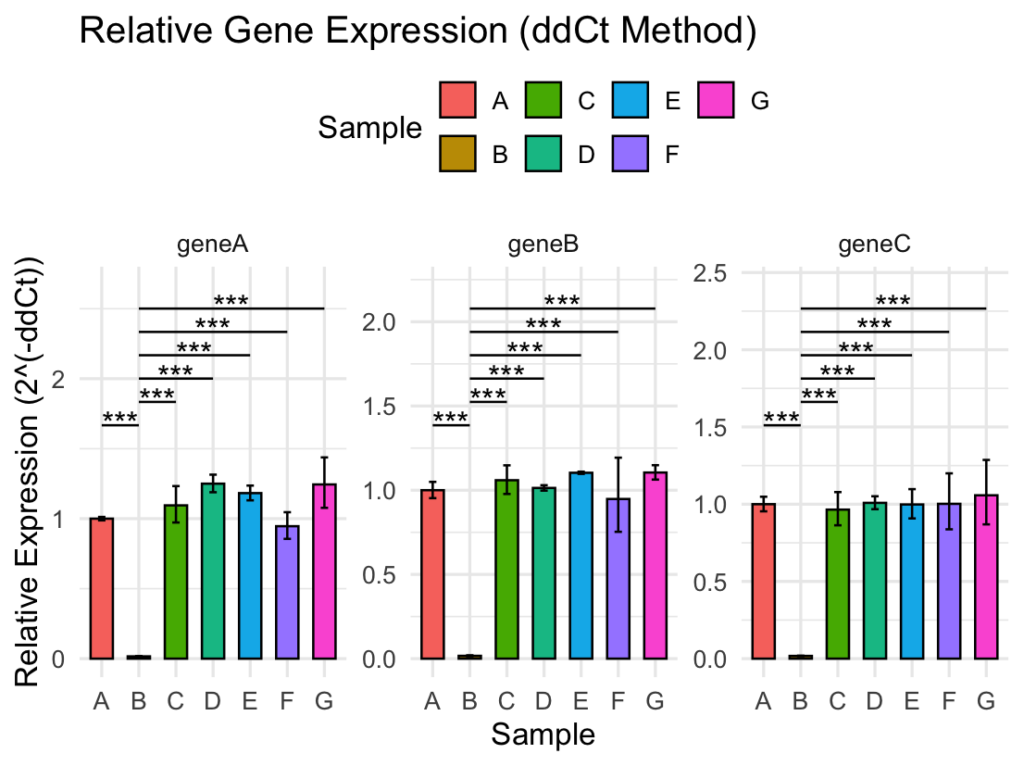
sample_colors <- setNames(hue_pal()(length(samples)), samples)
sample_colors <- setNames(hue_pal()(length(samples)), samples)hue_pal()は ggplot2 の内部で使われている 色のパレット関数です。hue_pal()(length(samples))で、サンプル数に応じた異なる色を生成します。setNames(..., samples)により、各サンプルに色名を割り当てた named ベクトルを作成することが出来ます。- この方法を用いれば、基本的にサンプル数がどれくらいであってもエラーが出ずにグラフを作成することが出来ます(色が似てしまうなどの不具合は出てきますが…)!
x 軸・y 軸・塗りの定義
ggplot(df, aes(x = factor(Sample, levels = samples), y = Relative_Expression, fill = Sample)) +- x 軸:Sample(levels = samples、と入力することで、最初に指定した順番にバーが表示されます。levels = samplesが無いと、アルファベット順にバーが表示されます。)
- y 軸:Relative_Expression(2^(-ddCt))
- 塗り色:fill = Sampleで、サンプルごとに、上の
sample_colorsで定義された色を使うようになります。
stat_summary()(棒グラフ)
stat_summary(
fun = function(x) exp(mean(log(x))),
geom = "bar",
color = "black",
width = 0.6,
position = position_dodge(0.7)
) +stat_summary()
ggplot2の関数で、データを要約統計値(mean, median, etc.)にしてプロットできる便利な関数です。- 通常の
geom_bar()とは異なり、「平均を取って棒グラフにする」用途に最適と思われたので、今回採用しました。
fun = function(x) exp(mean(log(x)))
- これは 幾何平均 (geometric mean) を計算する式です。
- 幾何平均は、「n個の数値を掛け合わせた積のn乗根を求めた平均値」です。
- log(x)で全ての数値のlogをとり、mean(log(x))で、ログスケールでの平均を取ります。
- これをexp()で、最後に指数を戻します。
2^(-ddCt)のように 比率データや 指数変換されたデータ(logスケール) は、
算術平均(いわゆる普通の平均)よりも幾何平均の方が中心傾向を適切に表すと言われています。- ちなみに、ExcelでもGEOMEAN()で幾何平均を求めることが出来ます。
geom = "bar"
- 結果として得られる幾何平均を 棒グラフで描くことを指定しています。
color = “black”
- 棒グラフの枠線の色を黒に設定します。
width = 0.6
- 棒の太さを指定(0~1の間で指定、1が最大)します。
position = position_dodge(0.7)
- 複数グループの棒グラフ(例:複数サンプル、複数ターゲット)を横に並べる(position_dodge)スクリプトです。
0.7は棒同士の間隔を決めるパラメータで、ここを変更することで間隔を狭めたり広げたりできます。
エラーバーの表示
stat_summary(
fun.data = function(x) {
m <- mean(log(x))
se <- sd(log(x)) / sqrt(length(x))
data.frame(
y = exp(m),
ymin = exp(m - se),
ymax = exp(m + se)
)
},
geom = "errorbar",
width = 0.2,
position = position_dodge(0.7),
color = "black"
) +stat_summary() を使うことで、生データから自動的に平均やSEを計算し、そのまま描画できます。
m <- mean(log(x))
xはそのバーに含まれるRelative_Expressionの数値ベクトルです(例:triplicateのデータ)。- logスケールでの平均値を計算しています(指数データなので log が適切と思われます)。
se <- sd(log(x)) / sqrt(length(x))
- 標準誤差(SE) = 標準偏差(SD)÷ √n
- この式では logスケールでの標準誤差を求めています。
なぜlogスケールで計算するのか?
2^-ddCt のような指数変化のあるデータは log変換すると正規分布に近づくため、統計的に扱いやすくなるからです。
MIQEガイドラインという、qPCRのpublicationのためのガイドラインでも、t検定・ANOVAなどの統計解析を適切に行うためには、log変換されたデータでの処理が望ましいとの記載があります。
data.frame(y = exp(m), ymin = exp(m - se), ymax = exp(m + se))
これがggplot に渡されるエラーバーの情報になります。
| 項目 | 内容 |
|---|---|
y | exp(m):棒グラフの高さ(幾何平均) |
ymin | exp(m – se):エラーバーの下端 |
ymax | exp(m + se):エラーバーの上端 |
- すべてlogスケールで計算したあとに元のスケール(exp)へ戻しています。
- こうすることで、比率データに適した対数空間での誤差の扱いが可能になります。
geom = "errorbar"
- 表示する形をエラーバー(縦の棒)に指定しています。
- 上下の値(
ymin,ymax)を使って、棒グラフの上に縦線を表示できるようになります。
width = 0.2
- **エラーバーの「横幅」**を指定します。
- 値を小さくすると細いエラーバー、値を大きくすると太めのエラーバーになります。
position = position_dodge(0.7)
- 複数のバーが 横並び(ドッジ)になっている場合、その位置に合わせてエラーバーも正しく横に配置する必要があります。
- この設定は、バーの設定と同じにする必要があります。でないと、エラーバーとバーのx軸位置がずれてしまいます!
- また、このスクリプトを入力しないと、エラーバーが全部中央に重なってしまうことになります。
color = "black"
- エラーバーの線の色を黒(black)に設定します。
グラフの統合
facet_wrap(~ Target, scales = "free_y") +これにより、各遺伝子ごと(Target)にRT-PCRの結果がグラフとして出力され、1つの図としてまとめられます。
scales = "free_y"によって、y軸はそれぞれ独立に調整されます。例えばgeneAの最大値が2、geneBの最大値が5だった場合、geneAのy軸は2より少し上までの表示になり、geneBのy軸は5より少し上の表示になります。
敢えてy軸を全てそろえたい場合には、scales = “free_y”をscales = “fixed”に変更してください。
色の設定
scale_fill_manual(values = sample_colors) +サンプル名と色の対応を手動で指定(上で作成した sample_colorsを元に)しています。
見栄えの調整
labs(
title = "Relative Gene Expression (ddCt Method)",
x = "Sample",
y = "Relative Expression (2^(-ddCt))"
) +
theme_minimal(base_size = 14) +
theme(legend.position = "top") +グラフの見栄えを整える、細かい調整です。
グラフのタイトル、x軸タイトル、y軸タイトルを設定します。
日本語だとバグ表示されてしまう(解決策はあるようですが、複雑なのでここでは述べません…)ので、Δは用いずddCtと表記しています。適宜変更してください。
theme_minimalでグラフのテーマを指定しています。詳しくは下記のリンクで説明されていました!

theme(legend.position = “top”)にて、凡例の位置を調整できます。デフォルトでは右側のようです。
“top”だと上側、”bottom”だと下側、”left”だと左側、となります。”none”で凡例を消すこともできます。
有意差の表記
geom_segment(
data = t_test_res_plot,
aes(x = x1, xend = x2, y = y_position, yend = y_position),
inherit.aes = FALSE,
color = "black",
linewidth = 0.5
) +
geom_text(
data = t_test_res_plot,
aes(x = x_mid, y = y_position + 0.01 * top_y, label = signif),
inherit.aes = FALSE,
size = 5
) +
geom_blank(data = y_limits, aes(x = 1, y = max_label_y))geom_segment()で、有意差があったペアの比較線を表示させています。
color = “black”で黒色に、linewidth = 0.5で線の太さを指定しています!
geom_text()で、有意差があったペアのアスタリスクを表示させます。
比較線の少し上を想定しており、y = y_position + 0.01 * top_yの”+0.01″を変更させることで、アスタリスクの位置を微調整できます。size = 5はアスタリスクの大きさです。
geom_blank(data = y_limits, aes(x = 1, y = max_label_y))については、細かいですが比較線・アスタリスクがグラフから見切れてしまうことを防ぐために設定しています。こちらを確認してください!
さいごに
以上にてスクリプトの説明は終了です!
RでPCRのグラフ作成を示したページが見当たらず、手探りでのスクリプト作成となりました。
想像以上に負荷がかかりましたが(笑)、なんとかスクリプトを自動化させて一般化させることができたのではと思います。
ここは違うだろなどご意見があれば、ぜひ頂きたいです!
また、今回はt検定を採用しましたが、それ以外の手法(Kruskal–Wallis test、Dunn検定など)については後日改めてスクリプト作成したいと思います!
スクリプト全景
library(dplyr)
library(ggplot2)
library(scales)
samples <- c("A", "B", "C", "D", "E", "F", "G")
target_genes <- c("geneA", "geneB", "geneC")
reference_gene <- "GAPDH"
replicates <- 3
set.seed(333)
df <- expand.grid(Sample = samples,
Target = c(reference_gene, target_genes),
Replicate = seq_len(replicates)) %>%
mutate(mean_Cq = case_when(
Target == reference_gene ~ 20,
Sample == "B" & Target != reference_gene ~ 28,
TRUE ~ 22
),
Cq = rnorm(n(), mean = mean_Cq, sd = 0.2)) %>%
dplyr::select(-mean_Cq)
head(df)
reference_df <- df %>%
filter(Target == reference_gene) %>%
dplyr::select(Sample, Replicate, reference_Cq = Cq)
df <- df %>%
filter(Target != reference_gene) %>%
left_join(reference_df, by = c("Sample", "Replicate")) %>%
mutate(dCt = Cq - reference_Cq)
baseline_df <- df %>%
filter(Sample == samples[1]) %>%
group_by(Target) %>%
summarise(baseline_dCt = mean(dCt, na.rm = TRUE), .groups = "drop")
df <- df %>%
left_join(baseline_df, by = "Target") %>%
mutate(
ddCt = dCt - baseline_dCt,
Relative_Expression = 2^(-ddCt)
)
t_test_res <- df %>%
group_by(Target) %>%
summarise(pmat = list(pairwise.t.test(Relative_Expression, Sample, p.adjust.method = "BH")$p.value), .groups = "drop") %>%
mutate(pmat = map(pmat, ~ as.data.frame(.x) %>%
tibble::rownames_to_column("Sample1"))) %>%
dplyr::select(Target, pmat) %>%
unnest(pmat) %>%
pivot_longer(cols = -c(Target, Sample1), names_to = "Sample2", values_to = "p_adj") %>%
filter(!is.na(p_adj) & p_adj < 0.05) %>%
mutate(signif = case_when(p_adj < 0.001 ~ "***", p_adj < 0.01 ~ "**", TRUE ~ "*"))
t_test_res_plot <- t_test_res %>%
group_by(Target) %>%
mutate(
top_y = max(df$Relative_Expression[df$Target == unique(Target)], na.rm = TRUE) * 1.05,
x1 = match(Sample1, samples),
x2 = match(Sample2, samples),
x_mid = (x1 + x2) / 2,
y_offset = row_number(),
y_position = top_y + (y_offset - 1) * 0.1 * top_y
) %>%
ungroup()
y_limits <- t_test_res_plot %>%
group_by(Target) %>%
summarise(max_label_y = max(y_position + 0.1 * top_y, na.rm = TRUE), .groups = "drop") %>%
mutate(Sample = samples[1])
sample_colors <- setNames(hue_pal()(length(samples)), samples)
ggplot(df, aes(x = factor(Sample, levels = samples), y = Relative_Expression, fill = Sample)) +
stat_summary(
fun = function(x) exp(mean(log(x))),
geom = "bar",
color = "black",
width = 0.6,
position = position_dodge(0.7)
) +
stat_summary(
fun.data = function(x) {
m <- mean(log(x))
se <- sd(log(x)) / sqrt(length(x))
data.frame(
y = exp(m),
ymin = exp(m - se),
ymax = exp(m + se)
)
},
geom = "errorbar",
width = 0.2,
position = position_dodge(0.7),
color = "black"
) +
facet_wrap(~ Target, scales = "free_y") +
scale_fill_manual(values = sample_colors) +
labs(
title = "Relative Gene Expression (ddCt Method)",
x = "Sample",
y = "Relative Expression (2^(-ddCt))"
) +
theme_minimal(base_size = 14) +
theme(legend.position = "top") +
geom_segment(
data = t_test_res_plot,
aes(x = x1, xend = x2, y = y_position, yend = y_position),
inherit.aes = FALSE,
color = "black",
linewidth = 0.5
) +
geom_text(
data = t_test_res_plot,
aes(x = x_mid, y = y_position + 0.01 * top_y, label = signif),
inherit.aes = FALSE,
size = 5
) +
geom_blank(data = y_limits, aes(x = 1, y = max_label_y))参考にしたブログ・HP・文献
- MIQEガイドライン:Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., Mueller, R., Nolan, T., Pfaffl, M. W., Shipley, G. L., Vandesompele, J., & Wittwer, C. T. (2009). The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clinical Chemistry, 55(4), 611–622. https://doi.org/10.1373/clinchem.2008.112797
- ggplot2のテーマはどれを使うべきか(https://qiita.com/ishiijunpei/items/23370f3c2e79f8cb72a0)


コメント